
This review summarizes evidence of dysregulated reward circuitry function in a range of neurodevelopmental and psychiatric disorders and genetic syndromes.
First, the contribution of identifying a core mechanistic process across disparate disorders to disease classification is discussed, followed by a review of the neurobiology of reward circuitry.
We next consider preclinical animal models and clinical evidence of reward-pathway dysfunction in a range of disorders, including psychiatric disorders (i.e., substance-use disorders, affective disorders, eating disorders, and obsessive compulsive disorders),
neurodevelopmental disorders (i.e., schizophrenia, attention-deficit/hyperactivity disorder, autism spectrum disorders, Tourette’s syndrome, conduct disorder/oppositional defiant disorder), and genetic syndromes (i.e., Fragile X syndrome, Prader-Willi syndrome, Williams syndrome, Angelman syndrome, and Rett syndrome). We also provide brief overviews of effective psychopharmacologic agents that have an effect on the dopamine system in these disorders.
This review concludes with methodological considerations for future research designed to more clearly probe reward-circuitry dysfunction, with the ultimate goal of improved intervention strategies.
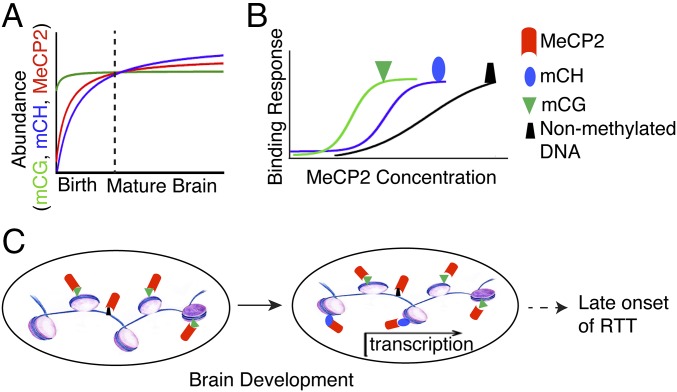
Rett syndrome (RTT) is a severe neurological disorder caused by mutations in the X-linked gene MECP2 (methyl-CpG-binding protein 2). Two decades of research have fostered the view that MeCP2 is a multifunctional chromatin protein that integrates diverse aspects of neuronal biology. More recently, studies have focused on specific RTT-associated mutations within the protein. This work has yielded molecular insights into the critical functions of MeCP2 that promise to simplify our understanding of RTT pathology.
Rett syndrome: a complex disorder with simple roots
OBJECTIVECurrent research suggests that the causes of autism spectrum disorders (ASD) are multifactorial and include both genetic and environmental factors. Several lines of evidence suggest that epigenetics also plays an important role in ASD etiology and that it might, in fact, integrate genetic and environmental influences to dysregulate neurodevelopmental processes.
The objective of this review is to illustrate how epigenetic modifications that are known to alter gene expression without changing primary DNA sequence may play a role in the etiology of ASD.METHODSIn this review, we summarize current knowledge about epigenetic modifications to genes and genomic regions possibly involved in the etiology of ASD.
RESULTSSeveral genetic syndromes comorbid with ASD, which include Rett, Fragile X, Prader-Willi, Angelman, and CHARGE (Coloboma of the eye, Heart defects, Atresia of the nasal choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness), all demonstrate dysregulation of epigenetic marks or epigenetic mechanisms.
We report also on genes or genomic regions exhibiting abnormal epigenetic regulation in association with either syndromic (15q11-13 maternal duplication) or nonsyndromic forms of ASD. Finally, we discuss the state of current knowledge regarding the etiologic role of environmental factors linked to both the development of ASD and epigenetic dysregulation.
CONCLUSIONSData reviewed in this article highlight a variety of situations in which epigenetic dysregulation is associated with the development of ASD, thereby supporting a role for epigenetics in the multifactorial etiologies of ASD.